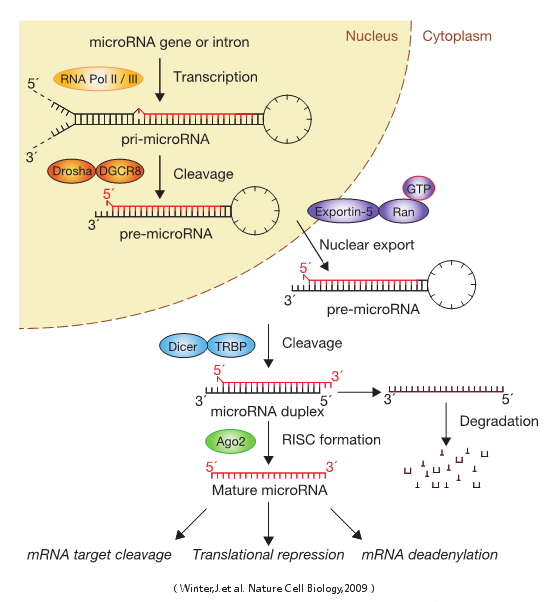
A microRNA (abbreviated miRNA) is a small non-coding RNA molecule (containing about 22 nucleotides) found in plants, animals, and some viruses, which functions in RNA silencing and post-transcriptional regulation of gene expression. Encoded by eukaryotic nuclear DNA in plants and animals and by viral DNA in certain viruses whose genome is based on DNA, miRNAs function via base-pairing with complementary sequences within mRNA molecules. As a result, these mRNA molecules are silenced by one or more of the following processes: 1) cleavage of the mRNA strand into two pieces, 2) destabilization of the mRNA through shortening of its poly(A) tail, and 3) less efficient translation of the mRNA into proteins by ribosomes.miRNAs resemble the small interfering RNAs (siRNAs) of the RNA interference (RNAi) pathway, except miRNAs derive from regions of RNA transcripts that fold back on themselves to form short hairpins, whereas siRNAs derive from longer regions of double-stranded RNA. The human genome may encode over 1000 miRNAs, which are abundant in many mammalian cell typesand appear to target about 60% of the genes of humans and other mammals.
Mammalian Gametogenesis
Gametogenesis is a biological process by which diploid or haploid precursor cells undergo cell division and differentiation to form mature haploid gametes. Depending on the biological life cycle of the organism, gametogenesis occurs by meiotic division of diploid gametocytes into various gametes, or by mitotic division of haploid gametogenous cells.
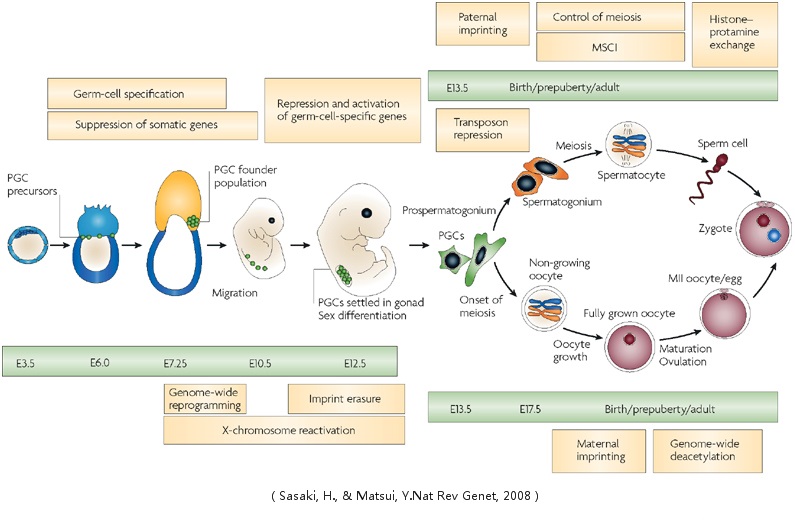
Spermatogenesis and Oogenesis
Spermatogenesis is the process in which spermatozoa are produced from male primordial germ cells by way of mitosis and meiosis. The initial cells in this pathway are called spermatogonia, which yield primary spermatocytes by mitosis. The primary spermatocyte divides meiotically (Meiosis I) into two secondary spermatocytes; each secondary spermatocyte divides into two spermatids by Meiosis II. These develop into mature spermatozoa, also known as sperm cells. In mammals, the first part of oogenesis starts in the germinal epithelium, which gives rise to the development of ovarian follicles, the functional unit of the ovary.Oogenesis consists of several sub-processes: oocytogenesis, ootidogenesis, and finally maturation to form an ovum (oogenesis proper). Folliculogenesis is a separate sub-process that accompanies and supports all three oogenetic sub-processes.
microRNA
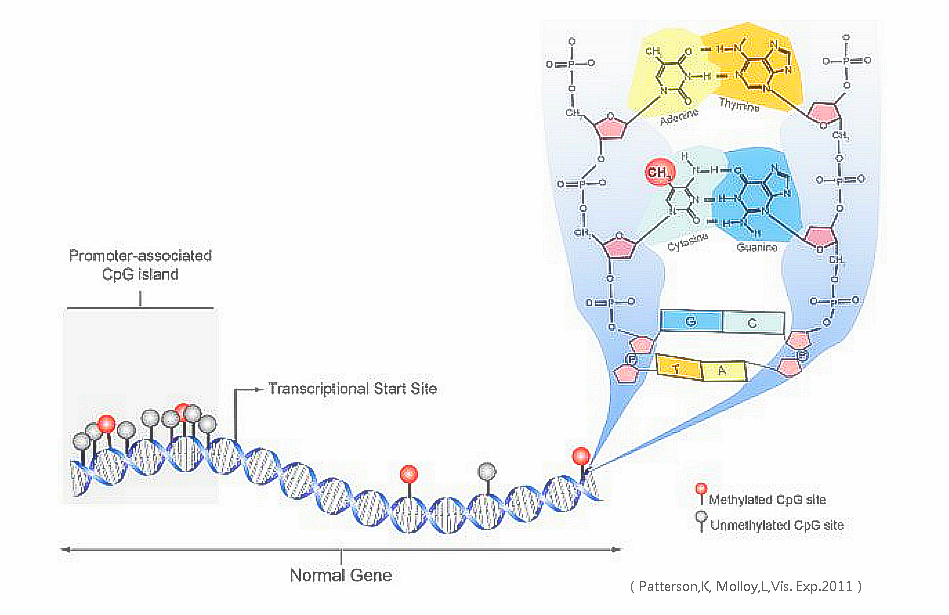
DNA methylation
Between 60% and 90% of all CpGs are methylated in mammals. Methylated C residues spontaneously deaminate to form T residues over time; hence CpG dinucleotides steadily deaminate to TpG dinucleotides, which is evidenced by the under-representation of CpG dinucleotides in the human genome. Unmethylated CpGs are often grouped in clusters called CpG islands, which are present in the 5' regulatory regions of many genes. In many disease processes, such as cancer, gene promoter CpG islands acquire abnormal hypermethylation, which results in transcriptional silencing that can be inherited by daughter cells following cell division. Alterations of DNA methylation have been recognized as an important component of cancer development. Hypomethylation, in general, arises earlier and is linked to chromosomal instability and loss of imprinting, whereas hypermethylation is associated with promoters and can arise secondary to gene (oncogene suppressor) silencing, but might be a target for epigenetic therapy. DNA methylation may affect the transcription of genes in two ways. First, the methylation of DNA itself may physically impede the binding of transcriptional proteins to the gene, and second, and likely more important, methylated DNA may be bound by proteins known as methyl-CpG-binding domain proteins (MBDs). MBD proteins then recruit additional proteins to the locus, such as histone deacetylases and other chromatin remodeling proteins that can modify histones, thereby forming compact, inactive chromatin, termed heterochromatin. This link between DNA methylation and chromatin structure is very important. In particular, loss of methyl-CpG-binding protein 2 (MeCP2) has been implicated in Rett syndrome; and methyl-CpG-binding domain protein 2 (MBD2) mediates the transcriptional silencing of hypermethylated genes in cancer. Research has suggested that long-term memory storage in humans may be regulated by DNA methylation. DNA methylation levels can be used to estimate age, forming an accurate biological clock in humans and chimpanzees.
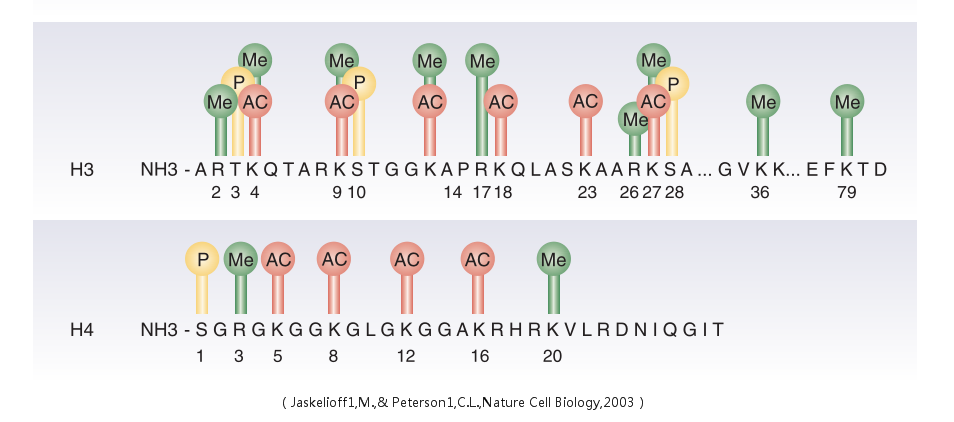
Histone modification
In biology, histones are highly alkaline proteins found in eukaryotic cell nuclei that package and order the DNA into structural units called nucleosomes.They are the chief protein components of chromatin, acting as spools around which DNA winds, and play a role in gene regulation. The addition of one, two or three methyl groups to lysine has little effect on the chemistry of the histone; methylation leaves the charge of the lysine intact and adds a minimal number of atoms so steric interactions are mostly unaffected. However, proteins containing Tudor, chromo or PHD domains, amongst others, can recognise lysine methylation with exquisite sensitivity and differentiate mono, di and tri-methyl lysine, to the extent that, for some lysines (e.g.: H4K20) mono, di and tri-methylation appear to have different meanings. Because of this, lysine methylation tends to be a very informative mark and dominates the known histone modification functions. What was said above of the chemistry of lysine methylation also applies to arginine methylation, and some protein domains—e.g., Tudor domains—can be specific for methyl arginine instead of methyl lysine. Arginine is known to be mono- or di-methylated, and methylation can be symmetric or asymmetric, potentially with different meanings. Addition of an acetyl group has a major chemical effect on lysine as it neutralises the positive charge. This reduces electrostatic attraction between the histone and the negatively charged DNA backbone, loosening the chromatin structure; highly acetylated histones form more accessible chromatin and tend to be associated with active transcription. Lysine acetylation appears to be less precise in meaning than methylation, in that histone acetyltransferases tend to act on more than one lysine; presumably this reflects the need to alter multiple lysines to have a significant effect on chromatin structure.